日外会誌. 126(5): 420-426, 2025
特集
医療機器開発と医工連携
3.医工連携の基礎知識―薬機法の承認取得とご遺体を使用した医療機器開発―
|
北海道大学病院消化器外科Ⅱ,先端医療技術教育研究開発センター 七戸 俊明 |
医療機器の開発には,薬機法(医薬品,医療機器等の品質,有効性及び安全性の確保等に関する法律)が定める人体へのリスクに応じた「クラス分類」の理解が不可欠である.医療機器はクラスⅠ~Ⅳに分類され,リスクが高いほど審査要件は厳格になる.クラスⅡ,Ⅲの医療機器で定められた認証基準に適合する場合は第三者認証(登録認証機関)により上市可能であるが,該当しない場合やクラスⅣの医療機器ではPMDA(医薬品医療機器総合機構)による審査と厚生労働大臣の承認が必要となる.
また,米国FDAや欧州連合のCEマークなど,各国で異なる認証制度が運用されており,国際展開にはそれぞれの制度を理解した開発戦略が求められる.
本稿では,新規医療機器の薬事承認制度,承認後の出口戦略(保険収載・市場導入)を概説するとともに,「医療機器の研究開発におけるカダバースタディーに関するガイダンス」を踏まえ,ご遺体を用いた医療機器開発についても述べる.
キーワード
薬機法, PMDA, クラス分類, 事前相談, カダバースタディー
I.はじめに
医療機器の開発では,技術中心のアプローチだけでなく,医療現場の課題を踏まえた医工連携が成功の鍵となる.近年,ロボティクスやAIの進展により,手術支援ロボットや画像診断支援などの高度な医療技術が実用化されているが,臨床で直面する課題を的確に捉え,新たな医療機器の開発につなげるためには,開発の初期段階からの外科医の関与が重要である.
本稿では,外科医が理解しておくべき医療機器の分類と承認制度の基本を解説するとともに,近年注目される医療機器開発におけるカダバースタディー(ご遺体を使用した研究開発)の意義と,その法的・倫理的留意点についても取り上げる.
II.医療機器の承認制度と国際的枠組み
1.薬機法の概要と目的
日本における医療機器の製造・販売・使用は,「医薬品,医療機器等の品質,有効性及び安全性の確保等に関する法律(以下,薬機法)」に基づく法的枠組みにより規制されている.薬機法は,医薬品と医療機器の双方について品質を確保し,国民の健康と生命を守ることを目的としているが,医療機器は,物理的あるいは機械的作用を主とする点で医薬品と異なり,独自の評価体系が採用されており,開発段階から製造,流通,市販後の安全管理に至るまで,医療機器のライフサイクル全体を対象とした包括的な規制が適用されている1).
2.医療機器の分類と審査制度
薬機法における医療機器の分類は,人体へのリスクの程度に応じてクラスⅠ~Ⅳに区分されており,機器の使用目的,作用機序,対象部位などに基づき判断される.また,これらの医療機器を製造・販売するには,分類に応じた厚生労働大臣の許可が必要である.具体的には,クラスⅠ(一般医療機器)には第三種,クラスⅡ(管理医療機器)には第二種,クラスⅢおよびクラスⅣ(高度管理医療機器)には第一種医療機器製造販売業許可が求められる2)(表1).
クラスⅠはリスクが低く,鋼製小物やガーゼなどが該当する.これらは製造販売業者による届出のみで市場に流通させることが可能である.クラスⅡは中程度のリスクを有し,内視鏡や超音波診断装置などがこれに含まれる.原則として登録認証機関(第三者認証機関)による認証を受ける必要があるが,該当製品が厚生労働省の定める認証基準に含まれない場合には,クラスⅢと同様にPMDA(Pharmaceuticals and Medical Devices Agency:医薬品医療機器総合機構)による技術審査と厚生労働大臣の承認が必要となる.クラスⅢおよびクラスⅣは,体内に長期間留置される医療機器や,生命維持に直結するような製品が該当し,人工呼吸器や人工関節などがクラスⅢに,心臓ペースメーカーやステントグラフトなどがクラスⅣに分類される.一部のクラスⅢ製品を除きこれらはPMDAの厳格な審査と厚生労働大臣の承認を経て上市される.
また,医療機器の申請区分による分類もある.既に製造販売の承認を受けている医療機器と構造,使用方法,効能,効果または性能が明らかに異なる医療機器を新規医療機器といい,承認済みの医療機器と同等な医療機器を後発医療機器という.また,新規医療機器,後発医療機器のいずれにも該当しないが,機能や構造に変更がある場合には,改良医療機器とみなされる.
近年,AIやICTを活用したソフトウェア単体の医療機器(SaMD:Software as a Medical Device)の開発が進んでいる.薬機法では2014年の改正以降,プログラム単体も医療機器として明確に規定され,他の医療機器と同様にクラスⅠ~Ⅳに分類される3).
クラスⅠには,リスクが極めて低く,生命・健康への影響が軽微と判断される一般医療機器が該当し,製造販売届出によって市場に供される.ただし,プログラムであっても医療目的を持たないものや,医療機器該当性を満たさないもの(例:電子問診ソフトや健康管理アプリなど)は,薬機法の規制対象外である.クラスⅡには視力検査支援ソフトや心電図解析補助ソフトなどがあり,認証基準に適合すれば登録認証機関による審査を経て上市される.クラスⅢおよびⅣには,AIを用いた診断支援プログラムや治療選択支援システムが該当し,PMDAの技術審査と厚生労働大臣の承認が必要となる.このように,プログラム医療機器も含めた全ての医療機器においては,初期段階で正確な分類と審査ルートを把握し,それに応じた計画立案やエビデンスの収集が,円滑な承認取得の鍵となる.
なお,これらの手続きを外科医自身が単独で担うことは現実的ではなく,医療機器製造販売業許可を持つ製販企業と連携して承認申請を進めるか,自らスタートアップを設立して対応する必要がある.また,AMED(国立研究開発法人日本医療研究開発機構)では,開発フェーズに応じた複数の支援事業を提供しており,開発早期からの活用が推奨される.
3.海外展開と各国の規制制度
日本の医療機器市場は規模に限りがあり,製品のライフサイクルも長いため,国際展開は実用化と収益化の鍵を握る.各国で医療機器の規制制度が異なることから,開発初期の段階で各国の制度を把握し,それに基づいた承認戦略を立てることが重要である.
米国での展開には,医療機器の規制を担当するFDA(Food and Drug Administration)の承認を得る必要がある4).医療機器の承認にはリスクに応じて,「510(k)」「De Novo」「PMA(Pre-Market Approval)」など複数のルートがあるが,クラスⅠまたはⅡに該当する新規医療機器で,既存製品との実質的同等性が認められる場合には,510(k)による申請が適用される.一方,同等製品が存在せず,かつ比較的リスクが低い場合には,De Novo申請を経てクラスⅠまたはⅡに再分類された上で,市場導入が可能となる.クラスⅢ以上に該当する高リスクの新規医療機器には,PMAによる科学的審査が求められ,有効性と安全性の十分なデータ提出が必要である.
欧州のEU加盟国での展開には,CEマーク(Conformité Européenne)の取得が必須である5).医療機器規則(MDR:Regulation (EU) 2017/745)に基づき,クラスⅠの一部は自己宣言で対応できるが,それ以上はNotified Body(認証機関)による審査が必須であり,技術文書や臨床評価報告書の整備が求められる.
アジアでの展開にも各国の承認が必要である.中国では,国家薬品監督管理局(National Medical Products Administration)が審査・承認を所轄し,現地代理人の指定や中国語での資料提出が義務付けられている.インドでは,CDSCO (Central Drugs Standard Control Organization)が医療機器の規制を担当しており,現地代理人の指定,輸入登録,インド規格への適合が求められる.
このように,各国がそれぞれ異なる制度を有しているため,医療機器を国際的に展開するには,制度的要件を早期に把握し,それに即した製品設計と申請資料の整備を進めることが不可欠である.なお,海外展開に当たっては,JETRO,PMDA,AMEDといった公的支援機関や,民間の専門支援機関の活用も有効である.

III.承認取得のための実務的手続き
1.PMDAの事前相談と審査
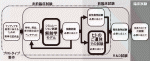
医療機器開発において,最初のマイルストーンは薬事承認の取得である(図1).医療機器の薬事承認を効率的かつ確実に取得するためには,PMDA(医薬品医療機器総合機構)が提供する各種相談・助言制度を,製品の開発フェーズに応じて適切に活用することが極めて重要である.これらの制度は,開発初期から申請準備,さらには承認申請直前の確認に至るまで,段階的に支援を提供する構成となっており,それぞれの目的と製品特性に応じて活用される6).
開発の初期段階では,「全般相談」が審査・承認制度の理解のための支援として設けられている.これは,PMDAの相談制度の全体像や活用方法について案内を受けるもので,特定の製品に対する技術的助言を行うものではない.制度の仕組みに不慣れなアカデミアやスタートアップにとって,初期段階の情報整理や戦略構築に有用である.なお,「全般相談」は無料で受けることができる.
より具体的な開発構想が固まり,非臨床試験の設計や治験の実施計画などを検討する段階では,「RS戦略相談(Regulatory Science戦略相談)」が活用される.この相談は有料ではあるが,承認取得に向けた製品特性に応じた戦略立案に対して,PMDAから実務的かつ科学的な助言を受けることができる制度である.アカデミアや中小企業でも利用が進んでおり,薬事戦略の明確化に資する制度として広く活用されている.
さらに,承認申請を目前に控えた段階では,「対面助言」が重要となる.この制度は,申請予定の資料や試験設計に関して,PMDAの審査担当者と直接意見交換を行う形式で実施される.新規性の高い製品や,既存の評価指標が適用しづらい機器においては,技術的・規制的な妥当性の事前すり合わせを行う手段として極めて有効である.
また,「対面助言」で得られた内容に基づいて資料修正等を行った後,その対応が適切であったかを確認する場として,「フォローアップ面談」が設けられている.これは対面助言の補完的制度であり,申請直前の段階で内容の最終確認や助言の再確認を目的として,申請者とPMDAが再度意見交換を行う.「フォローアップ面談」は無料で提供されており,形式的には新たな助言を行う場ではなく,あくまで進捗状況の確認と補足対応が中心となる.
このように,PMDAの相談制度は医療機器の開発フェーズごとに体系的に整備されており,段階的かつ戦略的にこれらを活用することが,開発初期の方向性の明確化,承認取得までの期間短縮,さらには開発コストの抑制にもつながる.特に医療機器開発に不慣れな外科医が主導するケースにおいては,早期の段階からこれらの制度を積極的に活用することが,実効性のある開発と社会実装への近道となる.
2.臨床試験の設計と代替手段の活用
新規薬剤の承認においては,ランダム化比較試験(RCT)を含むGCP(Good Clinical Practice:臨床試験の実施に関する基準)に準拠した臨床試験を通じて,有効性および安全性を科学的に検証することが原則とされている.これらの試験は,介入の効果を厳密かつ客観的に評価する標準的な方法として確立されており,審査当局による承認判断の根拠となる.一方,医療機器の開発においても原則として臨床試験による評価が求められるが,医薬品とは異なる構造的・操作的特性を有するため,同様の試験デザインを適用することが難しいケースが少なくない.たとえば,長期間使用に伴う安全性の検証,術者の習熟度による性能のばらつき,盲検化(ブラインド化)の技術的および倫理的な困難性,さらには侵襲性の高い機器に対するインフォームド・コンセントの取得の難しさなどが挙げられる.このような背景から,医療機器の承認審査においては,対象機器のリスク区分や使用目的に応じて,必ずしもGCP準拠の臨床試験に限らず,ベンチ試験(実験室での性能・安全性試験),動物試験,非臨床における機能評価などを組み合わせた柔軟な評価手法が認められている場合がある.たとえば,中リスクに分類されるクラスⅡ機器では,臨床使用成績調査や非ランダム化比較研究などにより,実地使用に基づいた有効性の補足的証拠が活用されることがある.特に低侵襲性を特徴とする機器や,術者による手技の違いがアウトカムに大きく影響する機器においては,「操作性」や「直感的な使いやすさ」といった定量化が難しい要素の評価が重要となる.このような場合,実臨床に近い条件下での評価手段として,手術に模した環境下での動物試験やカダバースタディー(ご遺体を用いた試験)などが代替的手段として注目されている.一方で,クラスⅢ以上の高リスク医療機器については,GCPに準拠した臨床試験の実施が原則とされ,特に新規性やリスクの高い機器では,効果と安全性を科学的に示す手段として不可欠である.これらの臨床試験は,被験者の人権保護と試験データの信頼性を確保するため,説明と同意の取得,倫理審査委員会の承認,適切なデータ管理体制など,国際的基準に則った厳格な管理が求められる.申請者は,これらの評価方法の選択にあたり,開発初期からPMDAとの協議を通じて,審査上受け入れ可能な試験デザインと評価手法を明確にしておく必要がある.
なお,承認申請から最終的な承認に至るまでの所要期間は,製品の分類や審査内容の複雑さに応じて変動するが,PMDAの統計によれば,標準的な審査から承認までの期間は約12カ月程度とされている7).

IV.承認後の出口戦略と保険収載の重要性
医療機器開発においては,薬事承認の取得後に実際の診療現場で使用されるための「出口戦略」が不可欠である.薬事承認のみでは医療機関での採用や臨床導入が保証されるわけではなく,とりわけ高額機器や操作に熟練を要する機器では,診療報酬制度における保険収載の有無が導入の可否を左右する決定的要素となる.保険収載は中央社会保険医療協議会(中医協)の審議を経て決定され,診療報酬体系における新規材料区分や技術料の設定,既存診療行為への包括,あるいは加算の適用といった形で評価される.これらの償還条件は,製品の普及可能性や医療現場での実装性に直結する.保険収載の審査では,薬事承認時のデータに加え,実臨床における有効性・安全性,さらには導入による医療的・経済的効果を定量的に示すことが求められる.再現性のある臨床データや医師による使用評価は重要なエビデンスとして審査に活用される.さらに,収載後の普及においては,専門医による実使用の評価や,臨床ガイドラインとの整合性の確保が普及の鍵となる.こうした信頼性の高いエビデンスの蓄積を通じて,医療機器としての有用性が医療界に認知され,新技術としての社会的地位が確立されていく.
このように,薬事承認はあくまでスタートラインに過ぎず,保険収載と実地エビデンスの整備を含む出口戦略の構築こそが,医療機器を臨床現場に届けるために不可欠なプロセスである.
V.医療機器の研究開発におけるカダバースタディー
医療機器の研究開発において,ご遺体(解剖体)を使用するカダバースタディーは,操作性や使用環境を再現可能な評価手段として注目されている.2025年に経済産業省とAMEDにより「医療機器の研究開発におけるカダバースタディーに関するガイダンス」が発出予定で,ガイダンスによりカダバースタディーの法的位置づけと実施手順が明確化される.カダバースタディーは,「死体解剖保存法」の定める解剖ではないが,商業的利用とみなされない公益性の下に,「人を対象とする生命科学・医学系研究に関する倫理指針」8),「臨床医学の教育及び研究における死体解剖のガイドライン」9)を遵守し,倫理審査委員会の承認,献体者ご本人と家族による書面での同意,個人情報の適切な管理,記録保存の義務など,厳格な倫理的要件を満たすことで実施可能である.
このような法的・倫理的枠組みを遵守した上で,カダバースタディーは医療機器の評価手法として重要な役割を担う.ベンチ試験や動物試験では再現が困難な人体の解剖学的構造の検証が必要な機器や,使用中の操作性が性能に直結する製品に対しては,実際の解剖構造を用いた評価により,設計段階での有用なフィードバックが得られる.特に新規の手術機器や埋め込み型デバイスの評価では,試作品のユーザビリティ検証や改良点の抽出に有効であり,使用成績調査や承認申請時の技術的根拠の補完として活用される可能性もある(図2).
ガイダンスの発出によって,これまで国内で基盤が無く海外で実施せざるをえなかったカダバースタディーが,今後,国内においても実施基盤が整うことが期待される.また,それによる医療機器開発の加速化にも期待したい.
VI.おわりに
本稿では,医工連携による医療機器開発において外科医が理解すべき薬機法の承認制度と,カダバースタディーの活用を含む実務的課題について述べた.今後は,アイデアを持つ外科医がこれらの制度を理解して医療機器開発に積極的に関わることで,革新的な医療機器が医療現場から数多く創出されることに期待したい.
利益相反:なし
文献
1) 厚生労働省ホームページ.医薬品,医療機器等の品質,有効性及び安全性の確保等に関する法律の概要.2025年5月8日. https://www.mhlw.go.jp/content/001424957.pdf
2) PMDAホームページ.承認審査関連業務 医療機器.2025年5月8日. https://www.pmda.go.jp/review-services/drug-reviews/about-reviews/devices/0028.html
3) PMDAホームページ.医療機器プログラム(SaMD)の審査ポイント.2025年5月8日. https://www.pmda.go.jp/review-services/drug-reviews/about-reviews/devices/0047.html
4) FDAホームページ.Device Advice:Comprehensive Regulatory Assistance.2025年5月8日. https://www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance
5) European Commissionホームページ.CE marking.2025年5月8日. https://single-market-economy.ec.europa.eu/single-market/ce-marking_en
6) PMDAホームページ.相談区分一覧表(医療機器・体外診断用医薬品).2025年5月8日. https://www.pmda.go.jp/review-services/f2f-pre/consultations/0019.html
7) PMDAホームページ.承認審査関連業務 新医療機器に係る承認審査の標準的プロセスにおけるタイムライン.2025年5月8日. https://www.pmda.go.jp/review-services/drug-reviews/about-reviews/devices/0022.html
8) 厚生労働省ホームページ.文部科学省,厚生労働省,経済産業省:人を対象とする生命科学・医学系研究に関する倫理指針.2025年5月8日. https://www.mhlw.go.jp/content/001077424.pdf
9) 日本外科学会ホームページ.日本外科学会・日本解剖学会:臨床医学の教育及び研究における死体解剖のガイドライン.2025年5月8日. https://jp.jssoc.or.jp/uploads/files/aboutus/guidelines/info20180406-01.pdf
10) AMEDホームページ.医療機器の研究開発マネジメントにおけるチェックポイント/ステージゲート.2025年5月8日. https://www.amed.go.jp/koubo/medical_device_check.html
11) ステファノス・ゼニオス,ジョシュ・マコーワー,ポール・ヨック:BIODESIGN バイオデザイン日本語版.薬事日報社,東京,2015.
PDFを閲覧するためには Adobe Reader が必要です。