日外会誌. 124(6): 485-491, 2023
特集
先天性嚢胞性肺疾患のup to date
4.分類に関する最新の知見2.病理診断の見地から
|
神奈川県立こども医療センター 病理診断科 田中 水緒 |
先天性嚢胞性肺疾患(congenital cystic lung disease, CCLD)の多くは肺炎を契機に切除されてきたが,近年では多くが胎児診断され精度の高い画像診断に基づいて適切な時期に手術が施行されるようになり,組織学的により詳細な検討が可能な検体の集積がなされるようになった.さらに最近の網羅的手法を含む遺伝子解析の著しい進歩により,一部のCCLDでは原因遺伝子が明らかにされつつある.これらの組織学的所見の集積と遺伝子異常の同定から,CCLDの疾患概念の整理・再編がされてきている.Congenital pulmonary airway malformation (CPAM)の5分類の各タイプはそれぞれ異なる発症機序が明らかにされつつあり,type 1およびtype 3の一部とtype 4についてはそれぞれKRAS遺伝子およびDICER1遺伝子を背景に前がん病変であることが示唆されている.また,気管支閉鎖症でしばしば観察される組織所見であるmicrocystic parenchymal maldevelopmentがCPAM type 2と診断されてきたものの多くや葉内・葉外肺分画症で認められ,肺の発生過程で気道閉鎖に引き続いて生じた共通の組織学的形態異常と認識された.CCLDの中には悪性腫瘍のポテンシャルを有する疾患も含まれ,正確な組織診断は臨床対応に肝要と考える.
キーワード
congenital pulmonary airway malformation, bronchial atresia, intrapulmonary/extrapulmonary sequestration, bronchogenic cyst
I.はじめに
先天性嚢胞性肺疾患(congenital cystic lung disease, CCLD)の病理診断はStockerが提唱した5分類(congenital pulmonary airway malformation, CPAM, type 0~4)1)が頻用されてきた.胎児期を含む画像診断技術の向上により,気管支の閉塞や分画肺の栄養血管などが正確に診断され,それに基づいた病理診断がなされるようになった.さらに多くの症例が胎児診断されるようになり,以前は反復する肺炎で発見されていたものが,計画的に切除され二次的所見の少ない組織学的評価良好な検体の病理所見が蓄積された.さらに,近年の著しい分子病理学的検査手法の進歩により,いくつかのCCLDの発症に関わる遺伝子異常が明らかにされつつある.一部のCCLDは腫瘍発生のポテンシャルをもつことも知られるようになった.そのような流れの中で,形態診断に加えて,遺伝子異常を含む病因に基づくCCLDの再分類が進んでいる.本稿では,いくつかのCCLDについて典型的な組織所見に最近の知見を加えて述べる.
II.CPAM type 0 (congenital acinar dysplasia, CAD)
CPAM type 0は肺の発生過程の偽腺管期における成熟停止とされており,非常に稀で絶対的に致死的な疾患である.Congenital acinar dysplasia (CAD)と同義である.肉眼的には充実性病変で,割面で微小嚢胞が散在して観察される.組織学的には小嚢胞は円柱上皮に覆われ,肺胞組織形成を欠く.本症の原因としてTBX4遺伝子およびFGF10遺伝子異常が報告されている2).
III.CPAM type 1および一部のCPAM type 3
CPAM type 1は肉眼的には1~数個の大型の嚢胞とその周囲の小型の嚢胞からなる.Stockerのオリジナルの分類では最大の嚢胞は2cm以上と定義されているが3),胎児診断され新生児期に切除された症例などでは2cmに満たない嚢胞のみからなる病変や,稀に小型の嚢胞のみからなるものもある.組織学的には嚢胞は線毛円柱上皮に覆われ,しばしば鋸歯状を示す(図1).上皮内の粘液産生細胞(mucogenic cell, MC)の集塊(図1インセット)はCPAM type 1に特徴的であるが,MCの出現頻度は症例で差があり,できるだけ多くの切片を作成しての観察が必要である.嚢胞上皮の扁平上皮仮性がときにみられる.嚢胞壁内には平滑筋束や,ときに軟骨組織を認める.
典型的なCPAM type 1の組織像を呈する病変のほとんどからKRAS遺伝子のコドン12のバリアントが検出されており4),一部の病変からはEGFR遺伝子の異常が指摘されている5).近年CPAM type 1の残存病変からの腺癌(主にmucinous adenocarcinoma)の発症が複数報告され,KRAS遺伝子を背景に前がん病変と認識されてきている6).同遺伝子異常はMC以外の嚢胞上皮からも検出され,残存のないよう切除されることが肝要である.一方,新生児・乳児期のCPAM type 1の病変の上皮はときにさまざまな程度の異型を示し,しばしば多くの分裂像が観察されるが,同時期においては腺癌の診断については慎重である必要がある.
CPAM type 3はStockerのオリジナルの分類では小型嚢胞からなり肉眼的に充実性病変と定義される.小嚢胞性病変でMCが観察されない一部のCPAM type 3と診断される病変からCPAM type 1と同様のKRASの変異が検出され,同病変の少なくとも一部はCPAM type 1と重複する疾患概念として捉えられている7).
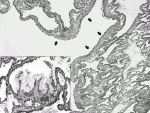
IV.CPAM type 4/ pleuropulmonay blastoma (PPB), type Ⅰ (purely cystic PPB)およびPPB type Ⅰr
CPAM type 4は通常胸膜直下の肺末梢部分に発症し,ときにポリープ状に突出する.肉眼的には薄い隔壁を伴う大型の嚢胞からなる.組織学的に嚢胞は肺胞上皮様の単層扁平上皮で覆われている(図2).嚢胞壁内は未熟な間葉系細胞が疎に分布している.
CPAM type 4はDICER1遺伝子の病的バリアントを背景にしばしばPPBと連続する病変であると認識されている.画像診断および肉眼診断ではCPAM type4とPPB type Ⅰ(pure cystic PPB)とは鑑別はときに困難である.CPAM type 4の組織診断においては,病変を可能な限り全て標本にした上で慎重に観察して,嚢胞の上皮下にcambium layerと呼ばれる芽球様細胞の集塊や隔壁内の横紋筋芽細胞,未熟な軟骨組織などPPBへの進展を示唆する所見の見落としのないようにする.
PPB typeⅠは充実成分を含むtypeⅡおよび充実性部分のみからなるtypeⅢに進展する.しかし,PPB typeⅠの一部で嚢胞構造を保ったまま壁内の芽球様細胞が消退するものが存在し,退縮型Ⅰ型 (typeⅠ regressed, typeⅠr)と称される8).乳児期以降に切除された検体で,線維化,ヘモジデリン沈着,組織球の集塊など退縮した腫瘍組織が考慮される所見を認めた際は,PPB typeⅠrを検討する.
CPAM type 4はDICER1関連腫瘍素因症候群で発症する特徴的な病変の一つである.DICER1関連腫瘍素因症候群はマイクロRNAのプロセッシングに必要な酵素をコードするDICER1遺伝子のヘテロ接合性の生殖細胞系列変異によって引き起こされ,さまざまな臓器の良・悪性腫瘍が発症することが知られている9).CPAM type 4の診断に際しては,可能な限りDICER1遺伝子異常の検索し,その上で好発腫瘍に対する適切なスクリーニングなど臨床対応が望まれる.

V.気管支閉鎖症 (bronchial atresia)
胎児期に起きた気管支の閉鎖に伴って生じる閉塞部位より末梢の肺実質の形成不全である.近年では画像診断の技術向上で術前に閉塞気管支が同定されている症例が多い.閉塞部位が中枢に近い症例では,肉眼的に切除検体の割面で閉塞部位が嚢状に拡張して粘液栓が充満しているものが観察されるが,より末梢で閉塞している症例では肉眼的に閉塞部位を同定するのは困難なことがある.閉塞部位より末梢では,微小な嚢胞状構造や気腫状変化がみられる.組織学的には拡張した気管支内に粘液栓が観察され,気管支閉鎖症の組織学的診断指標となる(図3).組織学的に閉塞部位を同定することはしばしば容易ではないが,閉塞部位近傍の気管支では変形した気管支軟骨や動脈の走行異常がみられる.閉塞部位より末梢の肺実質では線毛円柱上皮に覆われた小型の気管支様の嚢胞状構造がときに平滑筋束を伴って不規則に集簇するいわゆるmicrocystic parenchymal maldevelopmentの像が観察される.さらに末梢では気腫状変化を伴うことが多い.稀に病変内に横紋筋束が観察される(rhabdomyomatous dysplasia).Microcystic parenchymal maldevelopmentと気腫状変化はいずれも胎児期の肺発生過程で気道閉鎖に引き続いて生じた組織学的形態異常であると考えられており10),気腫状変化のみ観察される病変は先天性肺葉性肺気腫(congenital lobar emphysema)と診断される.
Microcystic parenchymal maldevelopmentは後述の肺分画症でもみられ,分画肺が本来の気道と交通していない状態で気管支閉鎖と同様の組織変化が生じたと考える.また,従来CPAM type 2と診断されてきた病変の多くがmicrocystic parenchymal maldevelopmentと同様と判断しうる所見を認め,さらにそのうち少なからず閉塞気管支を組織学的に同定しうる症例が含まれることが明らかになってきた.そのような中で,近年では気管支閉鎖症とCPAM type 2を同義する傾向にある11).現時点で,気管支閉塞以外にCPAM type 2の発症要因は遺伝子異常を含めて報告は見当たらない.

VI.葉内/葉外肺分画症(intrapulmonary/extrapulmonary sequestration, IPS/EPS)
肺分画症は肺の発生過程での過剰な肺芽が原因で生じた正常の気道とは交通のない気管支樹からなる肺組織であり,大動脈からの異常動脈に栄養されている.正常の肺と共通の胸膜に覆われている肺葉内肺分画症(IPS)と独立した胸膜を有する肺葉外肺分画症(EPS)がある.また,非常に稀に異常気管支が消化管と交通しているものもみられる(EPS with foregut communication).
IPSのほとんどは肺下葉にみられ,多くは大動脈からの異常動脈が流入している.異常動脈流入部付近にはリンパ節がしばしばみられ,分画肺の病変内に嚢状に拡張し,気管支軟骨や気管支腺を伴う気管支様構造と,肺門に向かって逆行性に分布する気管支樹が観察される(図4)12).正常気管支樹に通常欠陥はない.流入血管は弾性動脈である.分画肺の実質ではしばしばmicrocystic parenchymal maldevelopmentが観察されるが,CPAM type 2や気管支閉鎖症とのhybridとは診断しない.稀に病変内に横紋筋束が観察される(rhabdomyomatous dysplasia).実質での正常肺との境界は明瞭でないことが多い.流入動脈が筋性動脈であった場合,肺炎などに起因する反応性の異常血管である可能性が高く,IPSとは診断しない.一方,年長児で流入動脈が不明瞭になった症例においても,組織学的に末梢肺に弾性動脈を伴う異常気管支樹を同定することで肺葉内肺分画症の診断は可能である.
肺葉外肺分画症は固有の胸膜を有しており,診断は通常容易である.組織学的には肺葉内肺分画症と同様に拡張した弾性動脈の流入血管・気管支様構造・microcystic parenchymal maldevelopmentなどが観察される.
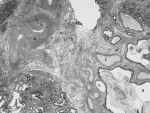
VII.気管支原性嚢胞(bronchogenic cyst)
気管支原性嚢胞は肺分画症と同様に過剰な肺芽が原因で生じ,ほとんどが中縦隔に,ときに肺門に接してみられる.肉眼的には,粘液性の内容物を含単嚢胞性病変で,通常は既存の気管・気管支・肺実質とは交通はない.大きさは4~5cmのものが多いが,年長児では10cm以上となることもある.組織学的には嚢胞壁は気管支上皮様の線毛円柱上皮に覆われ,壁内には軟骨,分泌腺,平滑筋束など気管支類似の構造が観察される(図5).類似の組織像を示す嚢胞性病変が腹腔内や皮下にも発生し,異所性肺芽から生じると考えられ同様に気管支原性嚢胞と診断する.

VIII.おわりに
CCLDの多くはそれぞれの疾患毎に特徴的な組織所見を有し,さらに画像所見や術中所見を合わせて鑑みることで,病理診断はそれほど困難でないことが多い.一方,一部のCCLDから悪性腫瘍の発生が明らかにされており,治療方法の選択肢に関わるため,組織学的検査に遺伝子検査の手法を加えて診断が求められる可能性がある.いずれにしても,精度の高い病理診断のために外科や放射線科など他科との十分な情報共有が望ましいと考える.
利益相反:なし
文献
1) Stocker J: Congenital pulmonary airway malformation:A new name for and an expanded classification of congenital cystic adenomatoid malformation of the lung. Symposium 24:Non- neoplastic Lung Disease Histopathology, 41: 424-430, 2002.
2) Karolak JA, Vincent M, Deutsch G, et al.: Complex Compound Inheritance of Lethal Lung Developmental Disorders Due to Disruption of the TBX-FGF Pathway. Am J Hum Genet, 104: 213-228, 2019.
3) Stocker JT, Madewell JE, Drake RM: Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol, 8: 155-171, 1977.
4) Nelson ND, Xu F, Chandrasekaran P, et al.: Defining the spatial landscape of KRAS mutated congenital pulmonary airway malformations:a distinct entity with a spectrum of histopathologic features. Mod Pathol, 35: 1870-1881, 2022.
5) Guo H, Cajaiba MM, Borys D, et al.: Expression of epidermal growth factor receptor, but not K-RAS mutations, is present in congenital cystic airway malformation/congenital pulmonary airway malformation. Hum Pathol, 38: 1772-1778, 2007.
6) Chang WC, Zhang YZ, Wolf JL, et al.: Mucinous adenocarcinoma arising in congenital pulmonary airway malformation:clinicopathological analysis of 37 cases. Histopathology, 78: 434-444, 2021.
7) Nelson ND, Xu F, Peranteau WH, et al.: Morphologic Features in Congenital Pulmonary Airway Malformations and Pulmonary Sequestrations Correlate With Mutation Status:A Mechanistic Approach to Classification. Am J Surg Pathol, 47: 568-579, 2023.
8) Hill DA, Jarzembowski JA, Priest JR, et al. : Type I pleuropulmonary:pathology and biology study of 51 cases from the i blastoma nternational pleuropulmonary blastoma registry. Am J Surg Pathol, 32: 282-295, 2008.
9) Gonzalez IA, Stewart DR, Schultz KAP, et al.: DICER1 tumor predisposition syndrome:an evolving story initiated with the pleuropulmonary blastoma. Mod Pathol, 35: 4-22, 2022.
10) Langston C: New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg, 12: 17-37, 2003.
11) Pogoriler J, Swarr D, Kreiger P, et al.: Congenital Cystic Lung Lesions:Redefining the Natural Distribution of Subtypes and Assessing the Risk of Malignancy. Am J Surg Pathol, 43: 47-55, 2019.
12) 石田 治雄:小児肺葉内肺分画症20例の検討-分画肺内気管支構造より.日胸外会誌,40:957-968,1992.
PDFを閲覧するためには Adobe Reader が必要です。