日外会誌. 123(1): 32-38, 2022
特集
Modern Surgeon-Scientistによる恒常性維持器官の外科研究
5.癌幹細胞の解析と新規治療の開発
|
東京医科歯科大学 医学部分子腫瘍医学 田中 真二 |
癌難治性の原因の一つは,その多様性・不均一性(heterogeneity)である.正常組織の多様性は自己複製能と多分化能を持つ幹細胞が担っているが,癌組織の中にも同様に「癌幹細胞」が存在することが明らかとなり,四半世紀以上が経過した.近年のシングルセル解析(omics),エピゲノム変動(bivalency),代謝リプログラミング(OXPHOS)などの研究によって癌幹細胞の本質的解明が進む中,生体内における免疫回避システムを巧妙に利用する癌幹細胞の強かな生存戦略も判ってきた.癌幹細胞を標的とする先端的研究を基盤として,様々な治療開発を進めている.
キーワード
癌幹細胞, エピゲノム変動, 代謝リプログラミング, 免疫微小環境, ゲノム編集
I.はじめに
癌治療には手術療法,化学療法,放射線療法,免疫療法など様々な手法が開発され,これらの治療が一旦奏功することも多い.しかしながら暫くdormantな状態が続いても,しばしば抵抗性をもった癌細胞集団へと変貌していき,浸潤・転移が進行して致死的となる.このような多様性・不均一性は「がん」の本質的特徴であり,難治性の原因となっている.癌の多様性・不均一性を説明するモデルの一つとして,元々起始となる特別な癌細胞が存在し,腫瘍形成はその階層性によって成立するという「階層性モデル」(hierarchy model)が唱えられた1).事実,生体内における腫瘍形成能は0.01%程度の癌細胞にしか認められない.正常組織の多様性は,自己複製能と多分化能を持つ幹細胞の非対称性分裂によって構成される階層システムが担うが,「階層性モデル」の起始癌細胞においても,不死化かつ非対称性分裂を示す幹細胞性が備わっていると推測されてきた.1994年ヒト急性骨髄性白血病細胞の中に幹細胞性を呈する起始細胞が内在することが証明されて以来,様々な組織由来の癌幹細胞が次々と見出され,現在に至っている.
癌幹細胞の同定には特定の幹細胞マーカーが用いられ,複数のマーカーの組み合わせにより癌細胞集団から単離することが可能である.しかし純化した癌幹細胞も非対称性分裂により,再び非癌幹細胞を生み出すため,癌幹細胞自体の特性を解析することは困難である.このジレンマを解決するには,癌肝細胞と非癌幹細胞を区別してモニタリングする手段が必須である.われわれは幹細胞のプロテアソーム非依存性に基づいて癌幹細胞を蛍光可視化し,膵癌,肝癌,大腸癌などの癌幹細胞をリアルタイムに可視化モニタリングするシステムを構築した(図1)2).癌幹細胞可視化システムを用いた解析により,癌幹細胞が非対称性分裂を繰り返して非癌幹細胞を生み出すプロセス,スフェア形成と細胞競合の動態,薬剤による細胞変化過程などをリアルタイムに解明した.さらに癌幹細胞には抗癌剤が無効であること,癌幹細胞特異的な薬剤スクリーニングが有効であること,癌幹細胞は1個でも生体内で不均一性腫瘍を形成し転移の根源となることを可視化した.近年シングルセル解析技術が開発され,癌幹細胞の遺伝的系統を追跡した結果,多様性を呈する癌幹細胞性クローン群が初期段階から存在し,そのまま維持されることが明らかとなった(ビッグバン・モデル)3).生体内における時間的(evolutional),空間的(spatial)解析手法も開発されており,癌幹細胞におけるエピゲノム変動,代謝リプログラミング,免疫微小環境の多層的解明が進んでいる.

II.癌幹細胞の可塑性とエピゲノム変動
幹細胞の非対称性分裂において遺伝子発現の可塑性を担うシステムの一つが,bivalentなクロマチン構造である(図2)4).クロマチンのbivalency(二価性)とは,ヒストンH3の活性型修飾である4番リジン残基のメチル化(H3K4me3)と,抑制型修飾である27番リジン残基のメチル化(H3K27me3)が同時に存在する状態であり,アクセルとブレーキを同時に掛けた空回りによって未分化性を保ちながら,分化シグナルなど環境の変化に対して速やかに呼応できるように備えていると考えられている.われわれは,可視化癌幹細胞の遺伝子発現がbivalentに制御されていることを見出しており,癌幹細胞の可塑性メカニズムの一つであることを示した1).腫瘍微小環境の変化に従って,H3K27me3脱メチル化による開放型クロマチン変動とDNA脱メチル化に伴う遺伝子発現亢進,或いはH3K4me3脱メチル化による閉鎖型クロマチン変動とDNAメチル化に伴う遺伝子発現抑制が誘導される.
このようなbivalencyは癌幹細胞の特徴であり,治療標的としても重要である.H3K27me3のメチル化を担う酵素EZH2(writer)に対して様々な阻害剤が開発されており,癌幹細胞の標的治療として有効性が示唆されている.H3K27me3の脱メチル化を担う酵素(eraser)にはJARID2,KDM6A,KDM6Bなどがある.膵癌の最も予後不良なサブタイプにはKDM6A変異が多いことが報告されており,われわれはゲノム編集を用いた解析を行い,KDM6A変異膵癌がヒストンアセチル化酵素とのCOMPASS様複合体形成不全によって幹細胞性,造腫瘍性を獲得することを見出し,ヒストン脱アセチル化酵素(HDAC)阻害薬に対して特異的な感受性を示すことを明らかにした5).
H3K4me3のメチル化酵素MLLは白血病など多くの腫瘍で変異を認め(writer),特異的阻害剤の開発により急性骨髄性白血病に対する臨床試験が進められている.またH3K4me3脱メチル化酵素としてKDM5A,KDM5Bや,H3K4me1,me2脱メチル化酵素としてLSD1/KDM1Aなどが報告されており(eraser),特にLSD1阻害剤の開発が進んでいる6).DNAメチル化酵素DNMTの阻害剤は急性骨髄性白血病等に対して承認されており,白血病幹細胞の自己複製能の阻害作用が確認されている.DNAメチル化阻害剤はゲノムに内在するトランスポゾン発現を誘導し免疫原性を高めることが知られているが,抑制型ヒストンマークであるH3K9me3のメチル化酵素SETDB1も同様の機序によって治療標的となることが最近報告された7).SETDB1増幅腫瘍では免疫チェックポイント阻害剤の効果が乏しいが,SETDB1を阻害するとヒストンアセチル化(H3K27ac)も亢進し,トランスポゾンが抗原として表出し,CD8+ T細胞の活性化が惹起される.DNAメチル化阻害剤とHDAC阻害剤の併用により,腫瘍免疫はさらに活性化されるため,癌幹細胞を枯渇させる複合免疫治療への応用が期待されている.

III.癌幹細胞と代謝リプログラミング
幹細胞の非対称性分裂において,特徴的なミトコンドリアの分配プロセスが報告されている8).新生ミトコンドリアは自己複製した幹性娘細胞へ選択的に割り当てられ,元々の古いミトコンドリアは分化した非幹性娘細胞へと分配される.この非対称性分配によって,幹細胞の系統は常に若々しいミトコンドリアが保たれる.ミトコンドリアによるエネルギー代謝は活性酸素を産生し,老化ミトコンドリアでは活性酸素濃度が高くなることが知られている8).われわれはミトコンドリア活性酸素が癌幹細胞で特異的に抑制されることを明らかにし,幹細胞性維持におけるミトコンドリア機能の重要性を示した9)10).
細胞のエネルギー代謝は解糖系(glycolysis)とミトコンドリアの酸化的リン酸化(oxidative phosphorylation; OXPHOS)が担っており,癌細胞は一般に解糖系に依存する(Warburg効果)(図3).一方,癌幹細胞ではOXPHOSによるエネルギー代謝へのリプログラミングが認められ11),基質となる脂質およびアミノ酸に対する依存性が報告されている12).JAK-STAT3経路活性化による脂肪酸酸化,NF-kB経路活性化による不飽和脂肪酸増加などが,癌幹細胞性の維持に不可欠であることが報告されている.脂肪酸に加えて,リン脂質のリモデリングも癌幹細胞の機能を制御している.腸管幹細胞でリン脂質生合成酵素LPCAT3をノックアウトすると,コレステロール生合成酵素の発現が誘導され,APC変異マウスでは腸管幹細胞が増殖し,腫瘍形成の促進による致死的効果をもたらす.LPCAT3ノックアウトの造腫瘍効果は,コレステロール合成阻害薬スタチンの薬理学的阻害によって抑制されることから,コレステロール合成の亢進が,LPCAT3欠失による癌幹細胞促進作用の原因であることが示された.脂肪酸およびコレステロール生合成は癌幹細胞の重要なドライバーであり,治療標的としても重要である.
癌細胞ではしばしばアミノ酸代謝も変化し,通常は非必須アミノ酸であるグルタミンが,生合成やエネルギー補給のために大量に消費されることが知られている12).腫瘍細胞におけるグルタミン代謝のリプログラミングは,山中4因子の一つであるMYCなど癌遺伝子によって誘導される.グルタミンは,特定の輸送体(ASCTファミリー)を介して細胞内に入り,グルタミン酸に変換され,続いてα-ケトグルタル酸に変換される.グルタミン酸は,活性酸素種(ROS)を除去する主要な物質であるグルタチオンの構成要素であり,致命的なDNA損傷・酸化損傷から細胞を保護する.グルタミン酸とシステインのトランスポーターxCTは,癌幹細胞マーカーCD44バリアントアイソフォーム(CD44v)と相互作用することが示されており,CD44vによって抗酸化物質グルタチオンの合成が亢進し,癌幹細胞の活性酸素に対する防御機構が強化される13).xCT阻害剤スルファサラジンは,癌幹細胞治療薬としての有効性が期待されている.さらにα-ケトグルタル酸は,ミトコンドリアのTCAサイクルの燃料となる必須の中間体であり,DNA脱メチル化酵素TETやヒストン脱メチル化酵素KDM4, KDM5ファミリーなどの補酵素でもある.多能性幹細胞ではLIF-STAT3シグナルによって,グルタミン酸からα-ケトグルタル酸への変換が促進し,DNA低メチル化によって可塑性が維持されている.腫瘍内ではT細胞よりも癌細胞が選択的にグルタミンを利用できるようにプログラムされていることが報告されており,免疫微小環境における代謝依存性を標的とする治療開発も進められている14).
IV.癌幹細胞と免疫微小環境
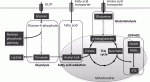
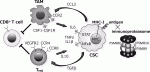
生体内における癌幹細胞の挙動について明らかにするため,われわれは可視化ヒト癌幹細胞を用いた転移モデルで解析している.肝癌幹細胞は高い転移能を呈し,転移巣の癌幹細胞周囲に宿主マクロファージが特異的に集簇しニッチを形成することを見出した9).癌幹細胞から発現するケモカインによってマクロファージが遊走することを明らかにして治療標的となる可能性を示した.マクロファージおよび制御性T細胞(Treg細胞)は,幹細胞ニッチの重要な構成要素である(図4)15).造血幹細胞,腸管幹細胞,乳腺幹細胞,毛包幹細胞などで,マクロファージとの機能的な相互作用があり,CCL2-CCR4シグナルなどのクロストークが作用する.Treg細胞も,様々な組織の幹細胞ニッチで確認されており,Treg細胞が産生するIL-10によって幹細胞の免疫優位性が維持される.癌幹細胞においても共通する宿主システムを操作し,自らに適した環境を形成することが認められている15).
抗原タンパクは,細胞内のプロテアソームでプロセッシングを受けてペプチドに分解され,トランスポーターTAPによって小胞体内に運搬されてMHCクラスI分子と結合する16).MHC-I-ペプチド複合体はゴルジ体を経て細胞表面に表出し,CD8T細胞により認識されて免疫反応が惹起される(図4).ES細胞におけるMHCの発現は低く分化に伴って陽性化するが,癌幹細胞でもMHCの発現低下が報告されている17).幹細胞性を誘導する癌遺伝子MYCもMHC発現を抑制することが認められており,抗原性低下による生体内での免疫回避メカニズムが示唆される.自己抗原は標準型プロテアソームによりプロセッシングされるが,癌抗原は主に免疫プロテアソーム(immunoproteasome)によってプロセッシングされる特徴を持っている.免疫プロテアソームは,標準型プロテアソームのコア部分の活性サブユニットPSMB5,PSMB6,PSMB7が,それぞれPSMB8,PSMB9,PSMB10に置き換わった構造である.興味深いことに,免疫プロテアソーム特異的サブユニットであるPSMB8,9,10の発現が低い腫瘍では,抗原性が抑制され免疫チェックポイント阻害剤も無効であることが臨床解析によって明らかとなった18).われわれは,可視化癌幹細胞においてPSMB8,9,10が特異的に抑制されていることを見出している.すなわち免疫プロテアソーム活性低下によりネオアンチゲンの提示を減少させ,宿主の免疫攻撃から癌幹細胞が巧妙に逃れている可能性が示唆される.生体内における癌幹細胞の免疫回避メカニズムの解明には,正常免疫モデルを用いた可視化癌幹細胞解析が必須である1).
V.おわりに
近年のオミックス解析技術の発展によって,様々な癌腫におけるサブタイプ分類が報告され,治療応用が期待されている.われわれは外科切除検体を用いた統合オミックス解析を行い,より精密な分子免疫サブタイプ分類を構築した19).同時にゲノム編集という優れた遺伝子デザイン技術が開発され,サブタイプ特異的なゲノム異常を再現することが可能となった20).当研究室では臨床解析を基盤として癌幹細胞のエピゲノム,代謝,免疫特異性を解明する多重ゲノム編集システムを構築し,正常免疫モデルを用いてどのサブタイプに,どのタイミングで,どのような組み合わせの治療が最適であるかを明確にする“CSC-targeted precision immunotherapy”の開発に取り組んでいる.シングルセル・オミックス解析と多重ゲノム編集というイノベーションの両輪を駆使することは,癌幹細胞を標的とした治療戦略へのさらなる展開を可能とする.精密な癌医療を外科手術に組み込み,根治的治癒を実現することがわれわれの使命である.
利益相反:なし
文献
1) Tanaka S: Cancer stem cells as therapeutic targets. In:Sherley JL (eds), Human Stem Cell Toxicity, Royal Society of Chemistry, London, pp280-294, 2016.
2) Adikrisna R, Tanaka S, Muramatsu S, et al.: Identification of pancreatic cancer stem cells and selective toxicity of chemotherapeutic agents. Gastroenterology, 143: 234-245, 2012.
3) Frank MH, Wilson BJ, Gold JS, et al.: Clinical implications of colorectal cancer stem cells in the age of single-cell omics and targeted therapies. Gastroenterology, 160: 1947-1960, 2021.
4) Blanco E, González-Ramírez M, Alcaine-Colet A, et al.: The Bivalent genome:characterization, structure, and regulation. Trends Genet, 36: 118-131, 2020.
5) Watanabe S,Shimada S, Akiyama Y, et al.: Loss of KDM6A characterizes a poor prognostic subtype of human pancreatic cancer and potentiates HDAC inhibitor lethality. Int J Cancer, 145: 192-205, 2019.
6) Mohammad HP, Barbash O, Creasy CL: Targeting epigenetic modifications in cancer therapy:Erasing the roadmap to cancer. Nat Med, 25, 403–418, 2019.
7) Griffin GK, Wu J, Iracheta-Vellve A, et al.: Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature, 595: 309-314, 2021.
8) Katajisto P, Döhla J, Chaffer CL, et al.: Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science, 348: 340-343, 2015.
9) Muramatsu S, Tanaka S, Mogushi K, et al.: Visualization of stem cell features in human hepatocellular carcinoma enlightened in vivo significance of tumor-host interaction and clinical implication. Hepatology, 58: 218-228, 2013.
10) Munakata K, Uemura M, Tanaka S, et al.: Cancer stem-like properties in colorectal cancer cells with low proteasome activity. Clin Cancer Res, 22: 5277-5286, 2016.
11) Bonnay F, Veloso A, Steinmann V, et al.: Oxidative metabolism drives immortalization of neural stem cells during tumorigenesis. Cell, 182: 1490-1507, 2020.
12) Jones CL, Inguva A, Jordan CT: Targeting energy metabolism in cancer stem cells:progress and challenges in leukemia and solid tumors. Cell Stem Cell, 28: 378-393, 2021.
13) Ishimoto T, Nagano O, Yae T, et al.: CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell, 19: 387-400, 2011.
14) Reinfeld BI, Madden MZ, Wolf MM, et al.: Cell-programmed nutrient partitioning in the tumour microenvironment. Nature, 593: 282-288, 2021.
15) Bayik D, Lathia JD: Cancer stem cell-immune cell crosstalk in tumour progression. Nat Rev Cancer, 21: 526-536, 2021.
16) Jhunjhunwala S, Hammer C, Delamarre L: Antigen presentation in cancer:insights into tumour immunogenicity and immune evasion. Nat Rev Cancer, 21: 298-312, 2021.
17) Pujadas E, Cordon-Cardo C: The human leukocyte antigen as a candidate tumor suppressor. Cancer Cell, 39: 586-589, 2021.
18) Kalaora S, Lee JS, Barnea E, et al.: Immunoproteasome expression is associated with better prognosis and response to checkpoint therapies in melanoma. Nat Commun, 11: 896, 2020.
19) Shimada S, Mogushi K, Akiyama Y, et al.: Comprehensive molecular and immunological characterization of hepatocellular carcinoma. EBioMedicine, 40: 457-470, 2019.
20) Tanaka S: Precision medicine based on surgical oncology in the era of genome-scale analysis and genome editing technology. Ann Gastroenterol Surg, 2: 106-115, 2018.
PDFを閲覧するためには Adobe Reader が必要です。